Test notebook for scirpy usage#
This notebook is the analog to the Colab created by Gregor S. for the demonstration of scirpy.datasets function usage:
https://colab.research.google.com/drive/15HIYGsSBHkDV399KSchv2QEyBzheL6fW?usp=sharing
import sys
sys.path.append('..')
import muon as mu
import numpy as np
import scanpy as sc
import scirpy as ir
# from iggytop.adapters.utils import AnnDataKG
# from biocypher.output.in_memory._airr_to_anndata import AIRRtoAnnDataKG as AnnDataKG
sc.set_figure_params(figsize=(4, 4))
sc.settings.verbosity = 2 # verbosity: errors (0), warnings (1), info (2), hints (3)
/home/docs/checkouts/readthedocs.org/user_builds/iggytop/envs/fruelingsputz/lib/python3.13/site-packages/muon/_core/preproc.py:31: FutureWarning: `__version__` is deprecated, use `importlib.metadata.version('scanpy')` instead
if Version(scanpy.__version__) < Version("1.10"):
Data import and preprocessing#
Loading example data and performing minimal preprocessing to get rid of “noise” data and further ignoring transcriptomic data preprocessing as we will not use it in this notebook. Incomplete cells (“orphan”) or potential doublets (“multichain”) are removed, which is common practice scAIRR-analysis. Refer to the data import tutorial and 3k T cell analysis tutorial for more comprehensive use-cases.
mdata = ir.datasets.wu2020_3k()
ir.pp.index_chains(mdata)
ir.tl.chain_qc(mdata)
mu.pp.filter_obs(mdata, "airr:chain_pairing", lambda x: ~np.isin(x, ["orphan VDJ", "orphan VJ", "multichain"]))
Downloading file 'wu2020_3k.h5mu' from 'https://exampledata.scverse.org/scirpy/wu2020_3k.h5mu' to '/home/docs/.cache/scirpy/0.24.0'.
0%| | 0.00/17.3M [00:00<?, ?B/s]
51%|███████████████████ | 8.89M/17.3M [00:00<00:00, 88.9MB/s]
0%| | 0.00/17.3M [00:00<?, ?B/s]
100%|█████████████████████████████████████| 17.3M/17.3M [00:00<00:00, 28.8GB/s]
/home/docs/checkouts/readthedocs.org/user_builds/iggytop/envs/fruelingsputz/lib/python3.13/site-packages/anndata/utils.py:362: ExperimentalFeatureWarning: Support for Awkward Arrays is currently experimental. Behavior may change in the future. Please report any issues you may encounter!
warnings.warn(msg, category, stacklevel=stacklevel)
/home/docs/checkouts/readthedocs.org/user_builds/iggytop/envs/fruelingsputz/lib/python3.13/site-packages/mudata/_core/mudata.py:1598: FutureWarning: From 0.4 .update() will not pull obs/var columns from individual modalities by default anymore. Set mudata.set_options(pull_on_update=False) to adopt the new behaviour, which will become the default. Use new pull_obs/pull_var and push_obs/push_var methods for more flexibility.
self._update_attr("var", axis=0, join_common=join_common)
Filtering chains...
Indexing VJ chains...
/home/docs/checkouts/readthedocs.org/user_builds/iggytop/envs/fruelingsputz/lib/python3.13/site-packages/mudata/_core/mudata.py:1461: FutureWarning: From 0.4 .update() will not pull obs/var columns from individual modalities by default anymore. Set mudata.set_options(pull_on_update=False) to adopt the new behaviour, which will become the default. Use new pull_obs/pull_var and push_obs/push_var methods for more flexibility.
self._update_attr("obs", axis=1, join_common=join_common)
Indexing VDJ chains...
build result array
Stored result in `mdata.obs["airr:receptor_type"]`.
Stored result in `mdata.obs["airr:receptor_subtype"]`.
Stored result in `mdata.obs["airr:chain_pairing"]`.
Clonotype clustering#
Clustering is performed here by calculating distances between individual sequences based on the BLOSUM62 matrix (metric = “alignment”) and choosing an appropriate cutoff. This will cluster similar TCR sequences with a distance below the chosen cutoff, which are likely to recognize similar antigens. For more information regarding different distance metrics and how receptor_arms and dual_ir affects the clustering algorithm please refer to the 3k T cell tutorial. However, the following three consecutive function calls are always required:
ir.pp.ir_dist(
mdata,
metric="alignment",
sequence="aa",
cutoff=15)
ir.tl.define_clonotype_clusters(mdata,
sequence="aa",
metric="alignment",
receptor_arms="all",
dual_ir="any")
#min_cells = 3 will only plot clonotype clusters with a size >= 3
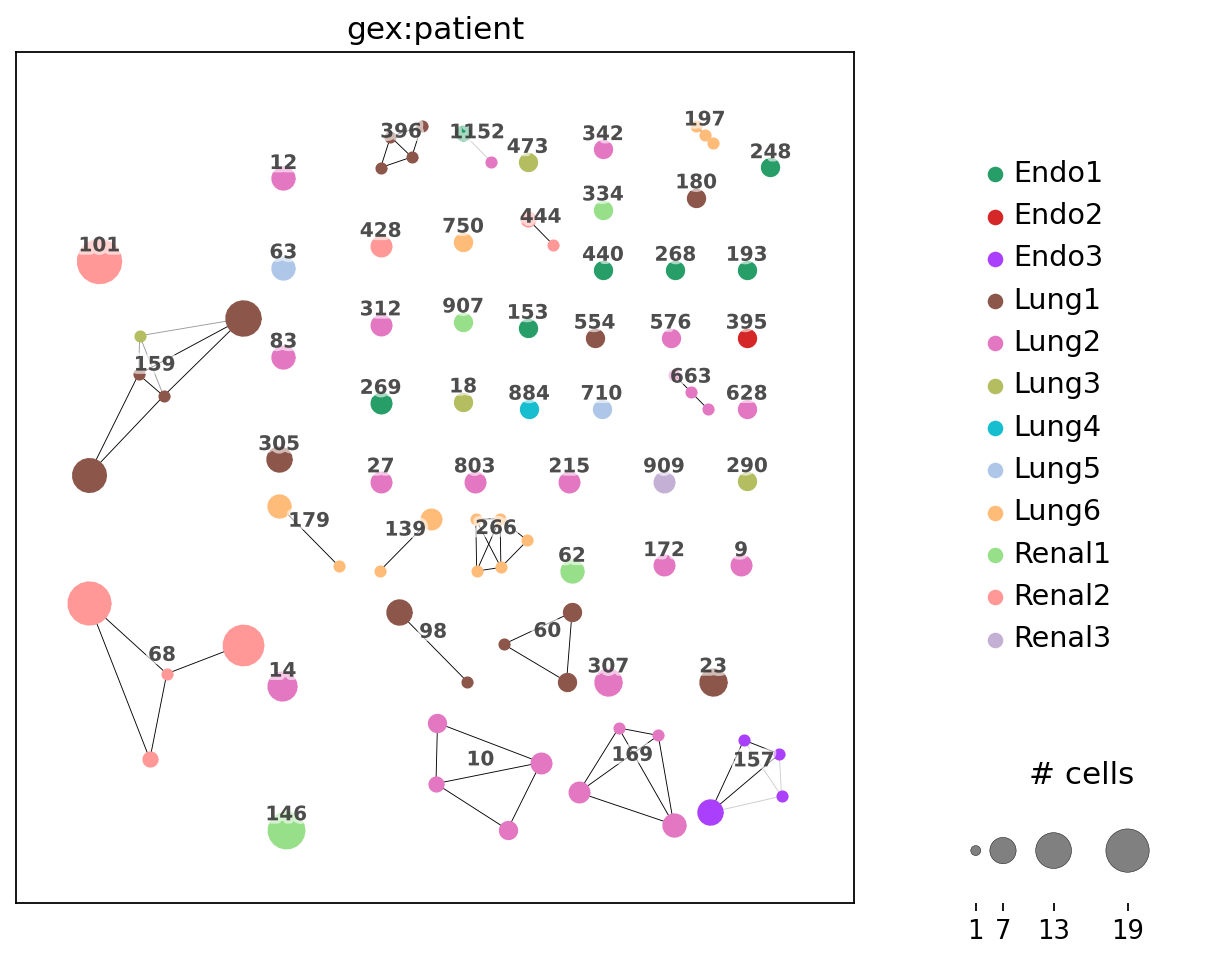
ir.tl.clonotype_network(mdata, min_cells=3, sequence="aa", metric="alignment")
_ = ir.pl.clonotype_network(mdata, color="gex:patient", label_fontsize=9, panel_size=(7, 7), base_size=20)
# save figure as a png
#plt.savefig("clonotype_network.png", bbox_inches="tight")
Computing sequence x sequence distance matrix for VJ sequences.
block size set to 50
Computing sequence x sequence distance matrix for VDJ sequences.
block size set to 50
/home/docs/checkouts/readthedocs.org/user_builds/iggytop/envs/fruelingsputz/lib/python3.13/site-packages/scirpy/ir_dist/metrics.py:98: FutureWarning: Input has data type uint8, but the output has been cast to float64. In the future, the output data type will match the input. To avoid this warning, set the `dtype` parameter to `None` to have the output dtype match the input, or set it to the desired output data type.
return triangular_matrix + triangular_matrix.T - scipy.sparse.diags(triangular_matrix.diagonal())
Initializing lookup tables.
Computing clonotype x clonotype distances.
Stored result in `mdata.obs["airr:cc_aa_alignment"]`.
Stored result in `mdata.obs["airr:cc_aa_alignment_size"]`.
/home/docs/checkouts/readthedocs.org/user_builds/iggytop/envs/fruelingsputz/lib/python3.13/site-packages/scirpy/pl/_clonotypes.py:179: FutureWarning: Use obsm (e.g. `k in adata.obsm` or `adata.obsm.keys() | {'u'}`) instead of AnnData.obsm_keys, AnnData.obsm_keys is deprecated and will be removed in the future.
if f"X_{basis}" not in params.adata.obsm_keys():
Load merged Iggytop data#
import scirpy as ir
from datetime import datetime
from scirpy.pp import index_chains
iggytop_tag = "data-2026.05.05.110331"
adata = ir.datasets.iggytop(tag=iggytop_tag)
Downloading data from 'https://github.com/biocypher/iggytop/releases/download/data-2026.05.05.110331/metadata.json' to file '/home/docs/.cache/scirpy/iggytop/a31892f7859278ab091910a296eb6dc2-metadata.json'.
SHA256 hash of downloaded file: 6c60d715cbd1c7b5e051641313ee6449bac509103c6b99e46d82c6359a240d02
Use this value as the 'known_hash' argument of 'pooch.retrieve' to ensure that the file hasn't changed if it is downloaded again in the future.
Downloading file 'deduplicated_anndata.h5ad' from 'https://github.com/biocypher/iggytop/releases/download/data-2026.05.05.110331/deduplicated_anndata.h5ad' to '/home/docs/.cache/scirpy/iggytop/2026.05.05.110331'.
index_chains(adata)
adata.uns["DB"] = {"name": "Iggytop", "date_downloaded": datetime.now().isoformat()}
# # store cache
# os.makedirs(os.path.dirname(os.path.abspath(cache_path)), exist_ok=True)
# adata.write_h5ad(cast(os.PathLike, cache_path))file
Filtering chains...
Indexing VJ chains...
Indexing VDJ chains...
build result array
ir.get.airr(adata, airr_variable = ["v_call","j_call", "junction_aa"], chain = ["VDJ_1"]).head(5)
| VDJ_1_v_call | VDJ_1_j_call | VDJ_1_junction_aa | |
|---|---|---|---|
| cell_id | |||
| 0_ITRAP | TRBV12-3 | TRBJ2-2 | CASSFSGNTGELFF |
| 1_ITRAP | TRBV27 | TRBJ2-2 | CASSPHFQVDTGELFF |
| 2_ITRAP | TRBV19 | TRBJ2-7 | CASSARSSYEQYF |
| 3_ITRAP | TRBV7-3 | TRBJ2-2 | CASSLGTSGGTGELFF |
| 4_ITRAP | TRBV7-9 | TRBJ2-7 | CASSSAAAYEQYF |
with ir.get.airr_context(adata, "j_call", "VDJ_1"):
print(adata.obs.columns)
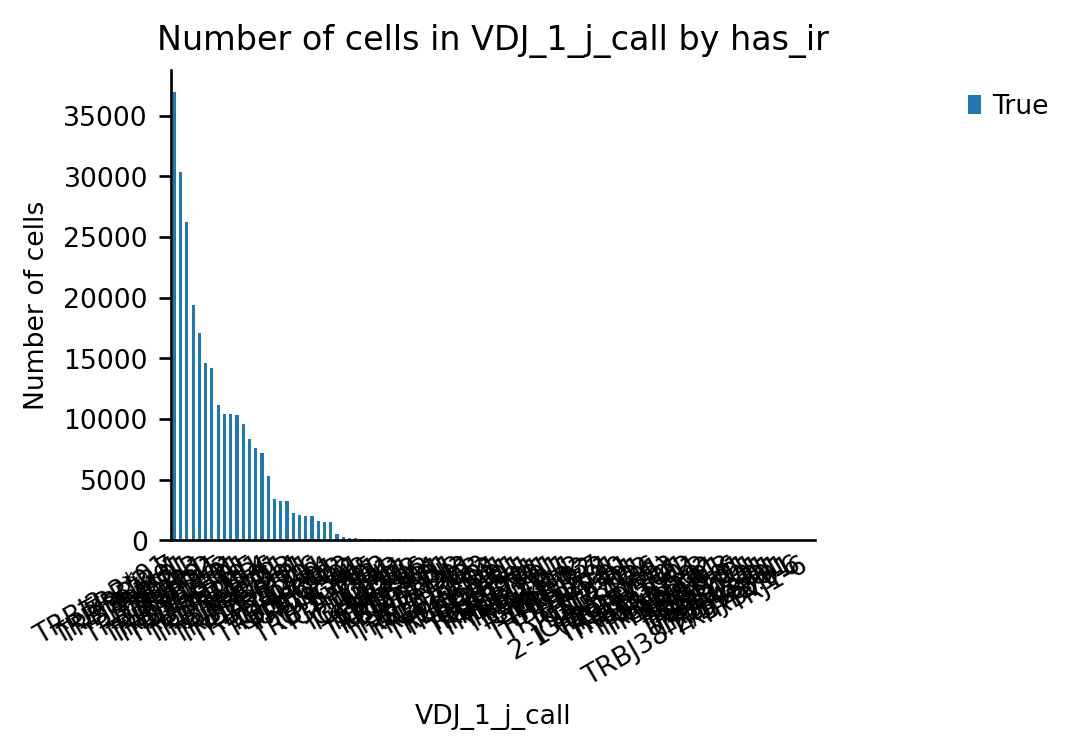
_= ir.pl.group_abundance(adata, groupby="VDJ_1_j_call", max_cols = 0)
# plt.xticks(rotation=50, ha='right')
Index(['MHC_gene_1', 'PMID', 'antigen_name', 'antigen_species',
'epitope_sequence', 'iedb_iri', 'source_organism', 'tissue',
'MHC_class', 'MHC_gene_2', 'source', 'VDJ_1_j_call'],
dtype='str')
ir.pp.ir_dist(mdata, adata, metric="fastalignment", sequence="aa", cutoff = 10)
Computing sequence x sequence distance matrix for VJ sequences.
block size set to 303
Computing sequence x sequence distance matrix for VDJ sequences.
block size set to 567
Next, cells between query and database are matched based on provided distance calculations and further conditions that can be specified in scirpy.tl.ir_query e.g. matching based on primary (dual_ir = “primary_only) CDRβ (receptor_arms = “VDJ”) sequences. For how Scirpy handles dual TCR please refer to the Immune receptor (IR) model
ir.tl.ir_query(
mdata,
adata,
metric="fastalignment",
sequence="aa",
receptor_arms="VDJ",
dual_ir="primary_only",
)
Initializing lookup tables.
Computing clonotype x clonotype distances.
Stored IR distance matrix in `adata.uns["ir_query_Iggytop_aa_fastalignment"]`.
Cells from the single-cell data are annotated with matched entries of the database based on selected conditions. Setting stategy = “most-frequent” reduces ambiguous assignments, but might reduce accuracy as well, as it will assign the most frequent match even if there are multiple different matches. Here, only one interesting column was included as annotation, although VDJdb contains much more information. This can be specified with the parameter include_ref_cols:
ir.tl.ir_query_annotate(
mdata,
adata,
metric="fastalignment",
sequence="aa",
strategy = "most-frequent",
include_ref_cols=["antigen_name"],
)
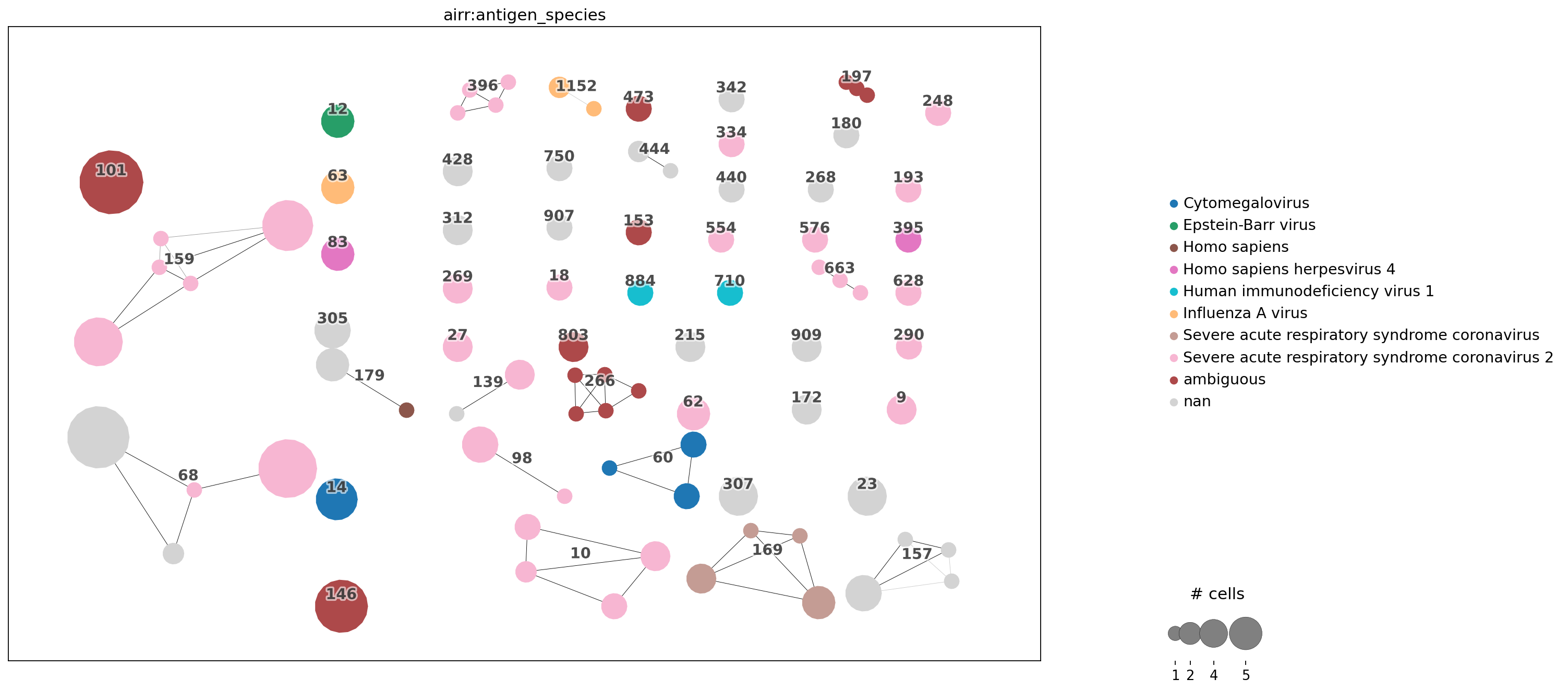
Stored result in `mdata.obs["airr:antigen_name"]`.
_=ir.pl.clonotype_network(mdata, color="airr:antigen_name", panel_size=(17, 10), show_legend=True)
/home/docs/checkouts/readthedocs.org/user_builds/iggytop/envs/fruelingsputz/lib/python3.13/site-packages/scirpy/pl/_clonotypes.py:179: FutureWarning: Use obsm (e.g. `k in adata.obsm` or `adata.obsm.keys() | {'u'}`) instead of AnnData.obsm_keys, AnnData.obsm_keys is deprecated and will be removed in the future.
if f"X_{basis}" not in params.adata.obsm_keys():

ir.tl.ir_query_annotate(
mdata,
adata,
metric="fastalignment",
sequence="aa",
strategy = "most-frequent",
include_ref_cols=["antigen_species"],
)
Stored result in `mdata.obs["airr:antigen_species"]`.
_=ir.pl.clonotype_network(mdata, color="airr:antigen_species", panel_size=(17, 10), show_legend=True)
/home/docs/checkouts/readthedocs.org/user_builds/iggytop/envs/fruelingsputz/lib/python3.13/site-packages/scirpy/pl/_clonotypes.py:179: FutureWarning: Use obsm (e.g. `k in adata.obsm` or `adata.obsm.keys() | {'u'}`) instead of AnnData.obsm_keys, AnnData.obsm_keys is deprecated and will be removed in the future.
if f"X_{basis}" not in params.adata.obsm_keys():
adata
AnnData object with n_obs × n_vars = 315055 × 0
obs: 'MHC_gene_1', 'PMID', 'antigen_name', 'antigen_species', 'epitope_sequence', 'iedb_iri', 'source_organism', 'tissue', 'MHC_class', 'MHC_gene_2', 'source'
uns: 'DB', 'chain_indices', 'iggytop_metadata'
obsm: 'airr', 'chain_indices'
ir.tl.ir_query_annotate(
mdata,
adata,
metric="fastalignment",
sequence="aa",
strategy = "most-frequent",
include_ref_cols=["source"],
)
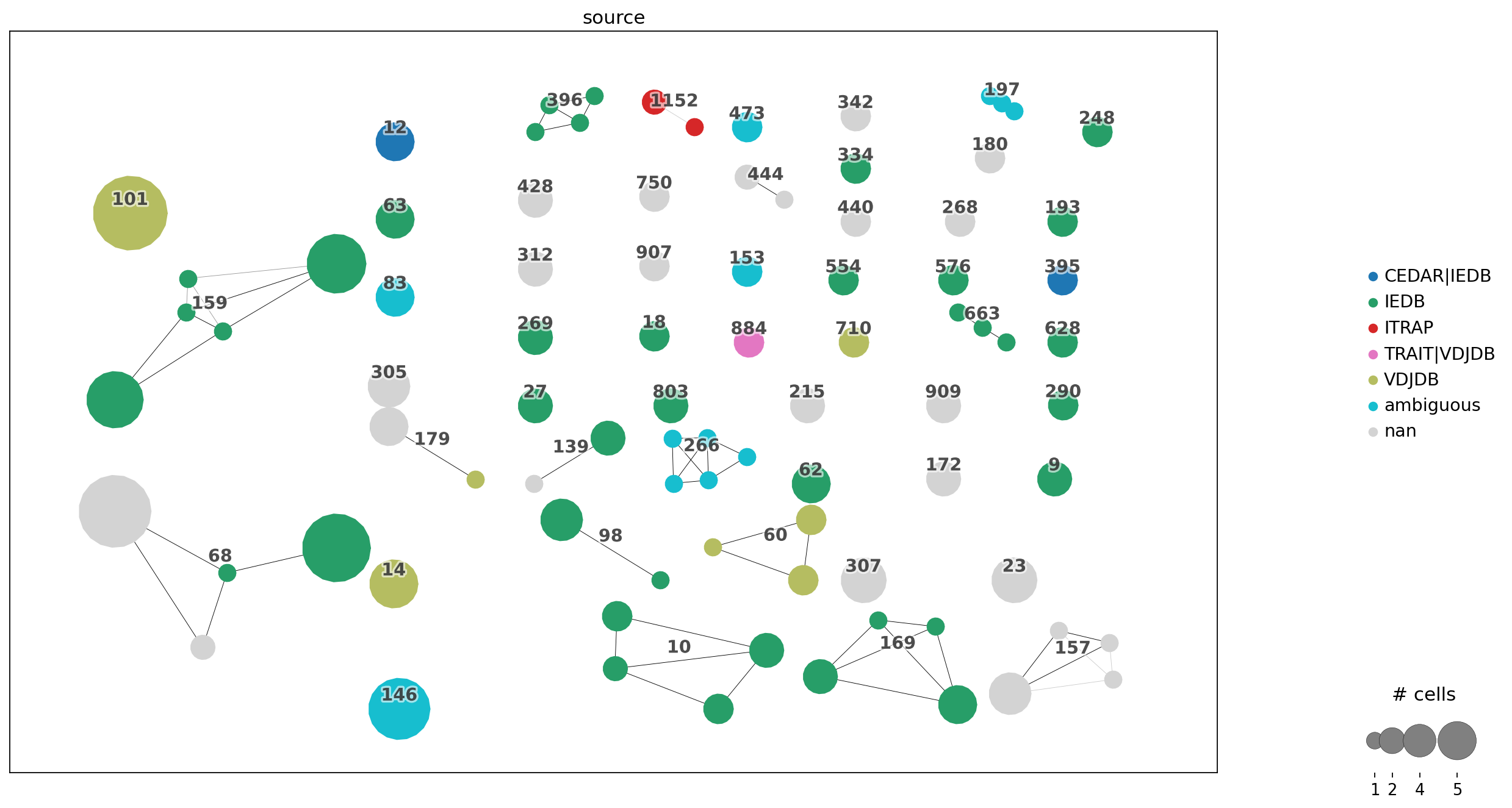
Stored result in `mdata.obs["airr:source"]`.
_=ir.pl.clonotype_network(mdata, color="source", panel_size=(17, 10), show_legend=True)
/home/docs/checkouts/readthedocs.org/user_builds/iggytop/envs/fruelingsputz/lib/python3.13/site-packages/scirpy/pl/_clonotypes.py:179: FutureWarning: Use obsm (e.g. `k in adata.obsm` or `adata.obsm.keys() | {'u'}`) instead of AnnData.obsm_keys, AnnData.obsm_keys is deprecated and will be removed in the future.
if f"X_{basis}" not in params.adata.obsm_keys():